
Help
1. General information
Homology modeling is one of the most successful computational approaches used for prediction of protein and RNA 3D structures. In the protein field, the concept of fix-backbone modeling of amino acid side chain conformations is successfully used. In contrast to that, there is no webserver tool dedicated for reliable modeling of nucleobase and nucleoside conformations onto fixed-backbone of RNA 3D structures. RNAfitme is designed to fill this gap. Our approach incorporates dedicated backbone-dependent conformer libraries. They encompass torsional angle distribution, investigated in a conformational space of high-resolution RNA 3D structures deposited in PDB. RNAfitme procedure finds the most promising 3D conformations where the number of steric conflicts is minimum. The web server can be also used to refine RNA 3D models generated by various computational methods even in top ranking 3D models assessed in RNA-Puzzles challenges, there is still a large number of atomic clashes.
The general idea of RNAfitme is presented on the following diagram:
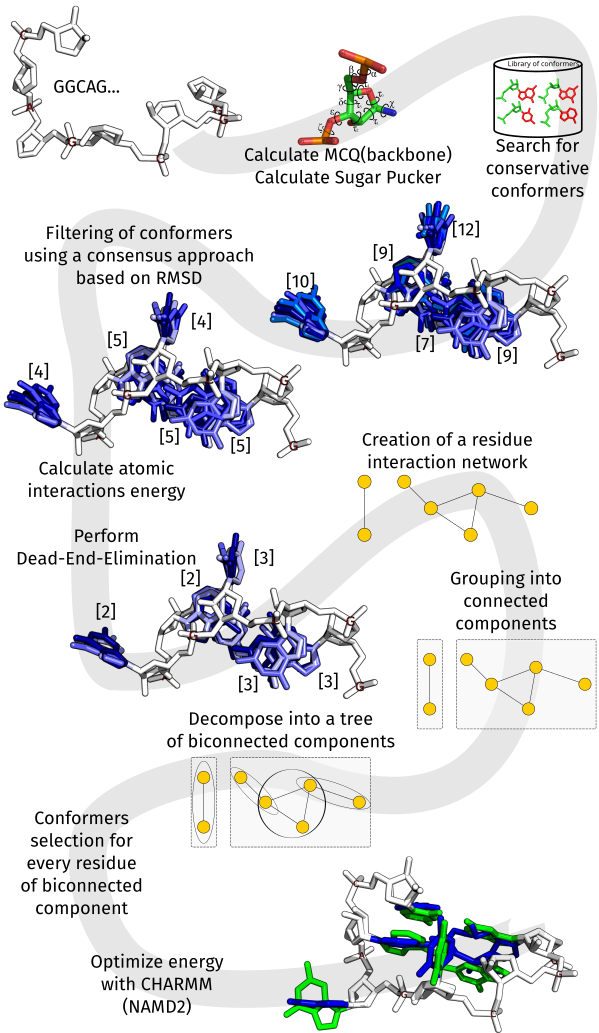
2. How to use RNAfitme
RNAfitme applies nucleotide conformer libraries within the RNA fixed-backbone concept. It runs according to the following scenarios (modes):
- in "nucleobase (re)placement", the coordinates of nucleobases are modeled based on fixed atom set including a backbone and a ribose ring of every nucleotide residue,
- in "nucleoside residue (re)placement", the coordinates of nucleoside residues are modeled based on fixed-backbone atom set of every nucleotide.
Step 1
In the first step of both scenarios, a user should upload the PDB file describing RNA 3D structure atom coordinates. The file can be uploaded either directly from a local disc (use "Browse" button to browse through the local repositories) or from Protein Data Bank. In the second case, a user should enter PDB identifier into the edit box, and click the "Get" button. The associated pdb file is automatically downloaded from PDB and made ready for processing by the application.
There are 4 example pdb files stored in the system and ready for processing. They enable an easy start with RNAfitme.
Uploaded data can be viewed in the textarea after clicking "Show file contents" button, and edited before further processing.
Step 2 (optional)
For homology modelling, the user introduces own sequence in FASTA format by clicking the checkbox connected with this option and pasting the sequence in the appearing textarea. Alternatively, the sequence can be uploaded directly from a local drive. The user can indicate residues which should be treated as rigid during prediction process. Atomic coordinates of such residues are not changed. In the input RNA sequence, every rigid residue is encoded using lowercase letter (i.e., 'a', 'c', 'g', 'u'). Such a residue must have the coordinates of all its atoms defined in the input structure. Atomic coordinates of all non-rigid nucleotide residues (provided in uppercase codes) are modeled during prediction process.
For multi-chain structure, the sequence should be a concatenation of sequences of considered chains without any delimiters such as ‐, & or other. For example, GCGC-GCGC should be introduced as GCGCGCGC.
Step 3
Next, the user should select the appropriate prediction mode (scenario), namely 'Nucleobase (re)placement' or 'Nucleoside residue (re)placement'. In the first mode, the uploaded RNA 3D structure should include the following atoms {O5', C5', C4', C3', O3', P, OP1, OP2, O4', C1', C2', O2'}. In the second mode, the following atoms should be defined in the structure {O5', C5', C4', C3', O3', P, OP1, OP2}. Atoms P, OP1, OP2 are optional for the first residue of the strand and can be generated automatically.
All atoms not required in the selected scenario are discarded. This especially concerns full-atom structures on input for which whole nucleobases or nucleosides are removed from further processing.
Step 4
Next, the user can decide what conformer library should be used during the prediction process. The system provides five libraries varying with methodologies applied during their construction, namely clustering method (i.e., neural gas and k-medoids), distance measure (i.e., euclidean and mcq), and data set of experimentally determined RNA 3D structures (i.e., a single RNA 3D structure - 23S rRNA, high-resolution RNA 3D structures (< 2.4 Å), all available RNA 3D structures deposited in RNA FRABASE).
Step 5 (optional)
The user can also decide to perform a final structure minimization using CHARMM force field within NAMD2 suite. In this step only the modeled atoms are being minimized. The rest of the RNA structure remains rigid.
Step 6 (optional)
Results can be also sent via email on user demand.
Step 7
To start the processing, the "Run" button should be clicked. This causes an immediate display of the results page where the following information is presented: a processing status of the current submission (i.e., enqueued, executed, processed), execution parameters set by the user, a log file where the prediction process is described in details, a visualization of predicted RNA 3D structure in JSmol (all (re)modeled atoms are green; the rigid part of RNA 3D structure uploaded by the user is blue). The predicted 3D model as well as the log file can be saved to a local drive. Moreover, the same input data can be processed again with the other set of execution parameters. If the submitted request cannot be executed in the moment of submission the user should remember the URL of current result page and visit this page later if possible to show the prediction output. To download the archive with prediction results, the user should check all checkboxes (under "3D model (PDB file) & Log file") associated with tasks of the interest. Finally, "Download selected results" button should be clicked.
3. System requirements
RNAfitme is designed to work with most of the available web browsers. The latest versions of browsers are strongly recommended.
| Browser | Recommended version |
|---|---|
| Google Chrome | ≥45 |
| Mozilla Firefox | ≥38 |
| Microsoft Edge | ≥12 |
| Internet Explorer | ≥10 |
| iOS | ≥9 |
| Safari | ≥9 |
| Android | ≥4.4 |
| Opera | ≥30 |
4. Citing RNAfitme
Any published work, which has made use of RNAfitme should cite the following paper:
M. Antczak, T. Zok, M. Osowiecki, M. Popenda, R.W. Adamiak, M. Szachniuk. RNAfitme: a webserver for modeling nucleobase and nucleoside residue conformation in fixed-backbone RNA structures, BMC Bioinformatics, 2018, 19, pp. 304 10.1186/s12859-018-2317-9.